AlphaFold3 跑一次很容易描述,可一旦反复跑就很昂贵。它的大部分耗时,都花在折叠与折叠之间并不变化的工作上:载入数据库、载入模型权重、对同一批同源序列一遍遍重搜。如果你的工作流是反复折叠同一组蛋白(扫突变、筛变体,或者一个多链复合物里大部分链固定、每次只改其中一条),那这笔固定开销,你每调用一次就要重付一次。
这篇文章讲的就是如何把它去掉。我会描述如何把 AlphaFold3(AF3)与 MMseqs2-GPU 组合成一个常驻、低延迟的折叠服务:对外是单个 HTTP 端点,在一台 8×80GB GPU 机器上供整个团队共享;并说明这套设计如何把最慢的 MSA 阶段,对重复输入压到近乎为零。讨论刻意保持与云厂商无关,因为同样的方案可以照搬到任意 8 卡 GPU 节点或 Kubernetes 在线推理平台上。服务代码已开源:v-shaoningli/alphafast-server(alphafast-serving 分支)。
关于范围。 这是一篇关于部署与服务层的文章:讲的是如何让折叠「反复调用也便宜」,而不是模型本身。我把 AF3 与 MMseqs2-GPU 当成既定前提,只关注它们外围的工程。整套部署建立在 AlphaFast(AF3 + MMseqs2-GPU)之上;模型与算法的功劳归 AlphaFold3(Abramson et al., 2024)与 MMseqs2(Steinegger & Söding, 2017)。完整致谢见文末。
这套做法归结为两步:一次性成本只付一次(而不是每次调用都重付),把重复的部分缓存下来。两步都做到,再折叠同一个靶点,就只剩推理这一步的开销了。
TL;DR
- 我把「每折叠一次就起一个一次性任务」换成了一个常驻的 HTTP 服务,从而消除冷启动、权重重载、数据库重载这些本会在每次调用上重复发生的开销。
- 五个加速叠在一起:数据库常驻显存(
gpuserver)、把比对放到 GPU(--prefilter-mode 3)、多链批量加多库并发搜索、模型权重常驻,以及序列级的 MSA 缓存。 - 一次冷请求约 230 秒(MSA 约 95 秒,推理约 134 秒);命中缓存的暖请求约 134 秒,因为序列一旦缓存,MSA 几乎免费。
- 最耗我时间的坑是把 gpuserver 的参数对齐:server 与 client 的
max-seqs、prefilter-mode、可见卡必须一致。
1. 为什么用服务,而不是一次性任务
AF3 跑一次结构预测,时间花在两段上。第一段是 MSA 与模板搜索,属 CPU 与 IO 密集型:它在几个大型序列库里搜同源序列:UniRef90(Suzek et al., 2015)、MGnify(Richardson et al., 2023)、一个精简版 BFD(Steinegger et al., 2019)和 UniProt(The UniProt Consortium, 2023);按传统做法(jackhmmer 或 HHblits),单条序列动辄几十分钟。第二段是 扩散推理,属 GPU 密集型:先载入几 GB 权重,再跑扩散采样。
问题在于,如果把每次折叠都当成一个一次性任务来跑,每次折叠都要从头重付同样的固定成本:拉起容器与环境、把几百 GB 的序列库从慢存储拷进本地或内存、把权重从磁盘载入显存。任务跑完即销毁,下一次再从零开始。
对于反复折叠同类输入的工作流,这些固定成本恰恰是大部分耗时。每次真正「新」的计算量很小,你却每次都要交满额的启动税。
常驻服务的做法,是把这些成本前置到启动阶段,于是每个请求只付它的增量成本。两种模式的对比相当鲜明:
| 成本项 | 一次性任务 | 服务 |
|---|---|---|
| 容器 / 环境 | 每次都付 | 只付一次 |
| 数据库载入 | 每次都付 | 只付一次(常驻内存 / 显存) |
| 模型权重载入 | 每次都付 | 只付一次(常驻显存) |
| MSA 搜索 | 每次都付 | 可缓存(相同序列免搜) |
2. 起点:AF3 加上 MMseqs2-GPU
如前所述,MSA 搜索是 AF3 的第一大瓶颈。最直接的杠杆,是把搜索引擎从 jackhmmer 换成 MMseqs2 的 GPU 版本,这通常能把 MSA 阶段提速一个数量级(社区基准约 68 倍)。
具体到这次部署,我直接构建在 AlphaFast(RomeroLab;Perry et al., 2026)之上。它是一个把 MMseqs2-GPU 集成进 AlphaFold3 数据流水线的开源衍生版,底层把 AlphaFold3(Google DeepMind)与 MMseqs2-GPU(Steinegger / Söding lab)组合在一起。本文的部署思路适用于任何「把 MSA 搬上 GPU」的 AF3 变体,但具体命令与行为以 AlphaFast 为准。
MMseqs2-GPU 给了我两样东西,正好对应下面前两层加速。第一样是工程上的:mmseqs gpuserver 把目标库常驻在 GPU 显存里,后续搜索直接连这个长生命周期进程,免去每次把库读进来的开销。
第二样才是加速本身。MMseqs2-GPU 的论文(Kallenborn et al., 2025)把搜索里恰好两个阶段搬到了 GPU 上。第一步是无间隔(gapless / ungapped)预过滤:把 query 的位置特异性打分矩阵(PSSM)映射到打分矩阵的列、参考序列映射到行,各行并行计算,并用 half2(在 32 位里打包两个 16 位浮点)来提升吞吐;这一步取代了 CPU 上基于 k-mer 或 SIMD 的预过滤。第二步是基于 PSSM 的带间隔(gapped)比对:对通过预过滤的序列做 Smith-Waterman-Gotoh 比对,用的是改造过、能直接在 PSSM 上运算的 CUDASW++4.0,以 wavefront 模式处理动态规划的依赖关系。
这两步具体跑哪一步,由 --prefilter-mode 决定,它有四个取值:0 是默认的 k-mer 预过滤(经典的 CPU 快速过滤);1 只做无间隔预过滤;2 不做预过滤,对每条参考序列都直接比对;3 则是无间隔加带间隔一起做。我用 --prefilter-mode 3,让上面两个 GPU 阶段都用上:无间隔预过滤负责筛掉大部分候选,带间隔比对再给留下来的序列打分,而不是让搜索回落到 CPU 的 k-mer 过滤。
3. 架构总览
展开细节之前,先看整体。以一台 8 卡 A100 机器为例,下面就是各卡最终的角色划分,也是通过了端到端集成测试的方案:
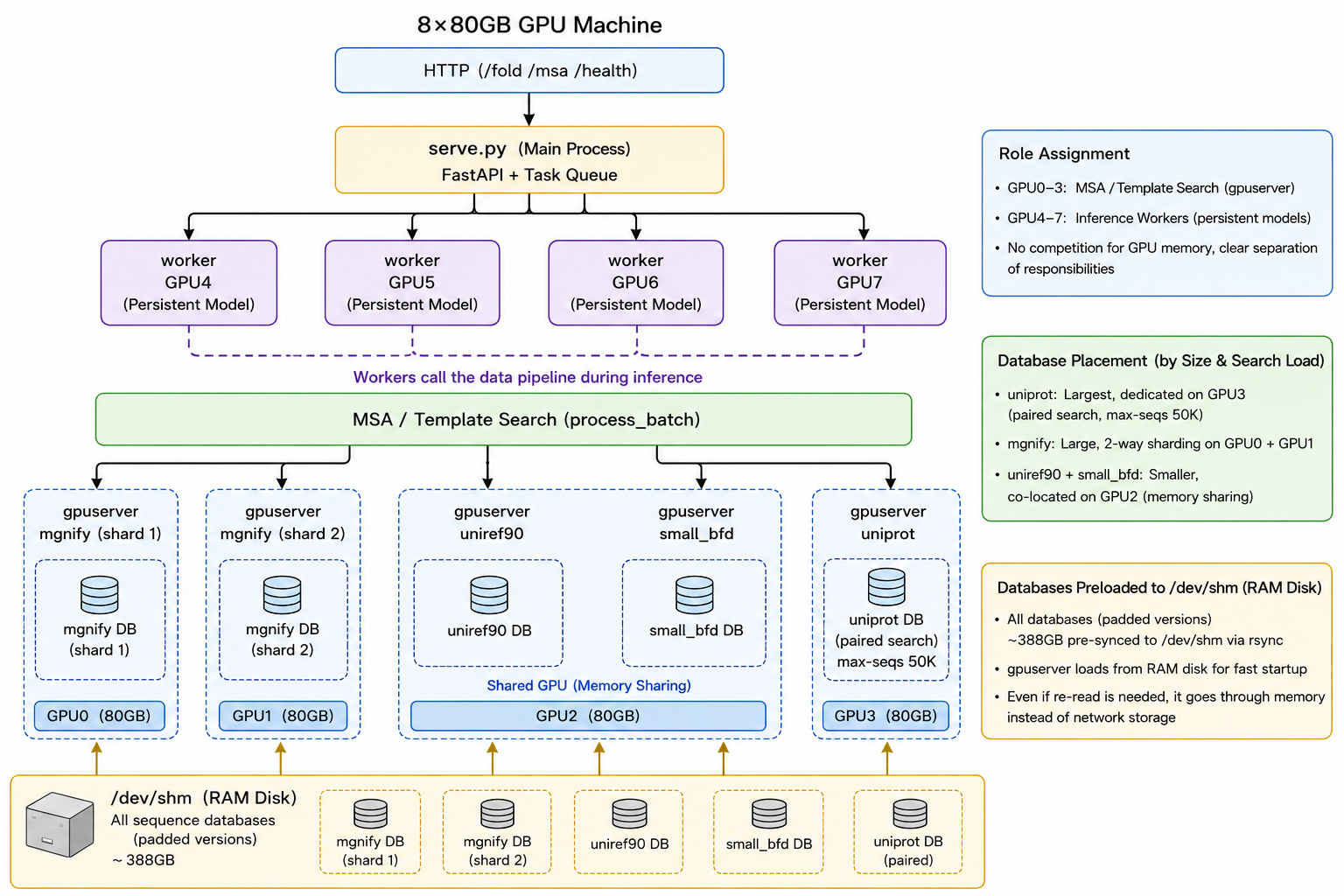
serve.py)把任务派发给 GPU4-7 上的四个推理 worker,每个常驻一份模型权重。MSA/模板搜索(process_batch)分散到 GPU0-3 上的四个 gpuserver,每个把一个库常驻在显存里;所有库预先落到 /dev/shm。(图片来源:本文,基于 AlphaFast。)这个布局背后有三点考虑。(1) 把 MSA 卡和推理卡分开。 GPU0-3 跑常驻的 MSA gpuserver,GPU4-7 跑常驻的推理 worker,两边互不抢显存。(2) 按库的大小和搜索量来摆放。 UniProt 最大(配对用,max-seqs 设到五万),让它独占一张卡;MGnify 也大,做两路分片占两张卡;UniRef90 与精简版 BFD 较小,合用一张卡。(3) 所有库先落到 /dev/shm(padded 版本合计约 388 GB),这样 gpuserver 从内存盘加载、启动更快,即便要回读也走内存而非网络存储。
4. 五层加速
加速不是单一技巧,而是五个各自独立的优化,每个消除一类不同的重复开销。其中四个都在打 MSA 这个瓶颈:把库常驻、把比对放到 GPU、把搜索批量加并发、以及缓存结果。第五个则让推理权重常驻。下面先讲三层 MSA 搜索优化,再讲那层推理优化,最后讲罩在整条 MSA 之上的缓存。
4.1 把库常驻显存(gpuserver)
普通的 mmseqs search --gpu 1 每次搜索都要先把目标库读进来,对几百 GB 的库来说光这一步就是分钟级。gpuserver 把它去掉:一个长生命周期进程把库一次性装进 VRAM,之后 mmseqs search --gpu-server 1 直接连它,成本就从「每次搜索都付」降成了「启动时付一次」。代价是显存被库长期占用,这正是要把 MSA 卡和推理卡分开的原因。
4.2 把比对放到 GPU(--prefilter-mode 3)
库常驻了,如果比对本身(尤其 gapped 比对)还悄悄回落到 CPU,也是白搭:结果是 CPU 满载、GPU 闲置。如第 2 节所述,--prefilter-mode 3 把 gapless 预过滤和 gapped 比对都留在 GPU 上。验证很简单:搜索时盯 nvidia-smi,对应库的那张卡应该贴近 100%。
4.3 批量处理链、并发搜索各库(process_batch)
最朴素的流水线对链、对库都串行:3 条链乘 4 个库就是 12 次串行搜索。两个维度都能压掉。链这个维度做批量:把一个复合物里所有链的唯一序列打包成每库一次搜索,让 GPU 一次并行处理多条 query。库这个维度做并发:四个库分布在不同 GPU 上,搜索同时发起,而且因为 gpuserver 让库常驻,并发不会因重复载入而 OOM。
一个值得点明的边界。 能并发的只有 GPU 搜索;紧随其后的
result2msa(把命中整理成 a3m)是 CPU 密集、并行扩展性很差。把四库搜索从串行改并发,冷 MSA 也只从约 100 秒降到约 95 秒(约 5%),因为真正的地板是 CPU 上的result2msa,并行化消不掉它;调不调它的线程数几乎没区别。能再往下压的只剩降max-seqs(牺牲 MSA 深度)或缓存。
4.4 让模型权重常驻
这是唯一一个关于推理、而不是 MSA 的优化。每个推理 worker 在启动时构建一次 ModelRunner,把权重载入它那张卡的显存并一直驻留,于是请求进来直接跑推理,省掉每次几 GB 的权重载入。换算很简单:N 张推理卡 = N 个 worker = N 份常驻权重 = 可同时进行 N 个折叠。
4.5 按序列缓存成品 MSA
即便搜索很快,对反复出现的序列(一批相关输入里共享的那些链)重搜仍是重复劳动。缓存能消掉它,唯一要定的是粒度。方案 A 缓存各库的原始 a3m(键 = 库 + 序列),命中后还得做解析与跨库去重,也就是 result2msa 之外那部分 CPU 工作。方案 B 缓存成品 MSA(键 = 序列):把去重后的 unpaired/paired MSA 连同模板存成一份 JSON,命中后原样灌回该链,搜索、解析、去重、模板一并跳过。
对配置固定的在线服务(库集合、max-seqs、去重逻辑都不变),方案 B 更对路:「序列对应它的成品 MSA」是一个稳定、可缓存的产物,方案 A 那种「可重新组合的中间产物」的灵活性在这里没有用武之地。差距很大:
| 冷 | 暖(命中) | |
|---|---|---|
| 方案 A | ~95 s | ~1.3 s(还要解析 + 去重) |
| 方案 B | ~95 s | ~0.0 到 0.1 s(整条链跳过) |
实现几乎是免费的。AF3 的输入 JSON 里,每条链都能预填 unpairedMsa、pairedMsa、templates,一旦预填,数据流水线就跳过这条链的搜索。所以方案 B 不动流水线,只要在请求进入流水线前把命中的链按缓存灌好,天然复用 AF3 自带的「给了 MSA 就不搜」逻辑。唯一的提醒:配置一变(换库、改 max-seqs、改去重)缓存就失效、得清空,两种方案都一样。
这也正是本节的落点:缓存并不降低冷成本,而是绕过它。暖请求整段跳过 MSA,但冷请求仍被 CPU 上的 result2msa 钉在约 80 到 90 秒。这块地板就是本节一切的天花板:上面每一层要么避开 MSA、要么把它搬上 GPU,而剩在 CPU 上的,就是你在不牺牲 MSA 深度的前提下再也压不动的部分。
5. 服务层:serve.py 怎么设计
本节的完整代码在 GitHub 上:v-shaoningli/alphafast-server(alphafast-serving 分支)。骨架刻意保持简单:一个 FastAPI 主进程,每张推理卡一个 worker 子进程,中间用一个任务队列解耦。一个请求的生命周期大致是这样:
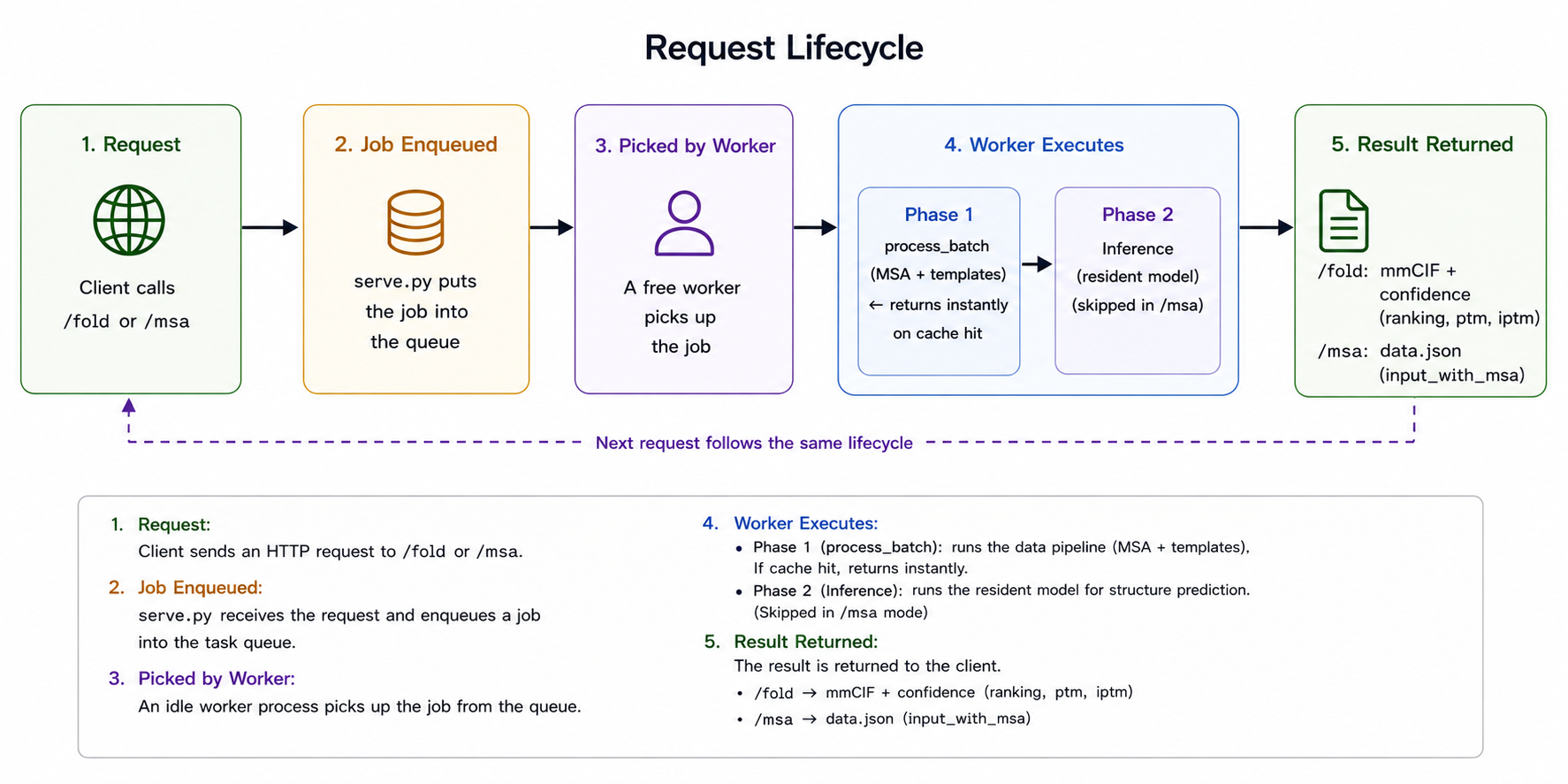
serve.py 里的生命周期。/fold 或 /msa 调用入队、被空闲 worker 取走,worker 先跑 phase 1(数据流水线的 process_batch,负责 MSA 与模板,命中缓存则秒回),再跑 phase 2(常驻模型推理,/msa 模式跳过),最后返回结构加置信度,或返回 MSA。几个接口都是围绕实际使用习惯设计的。/fold 做完整折叠,返回结构(mmCIF)加置信度(ranking、ptm、iptm)。/msa 只搜 MSA 与模板,返回一份可复用的 data.json,留给「只想要 MSA、暂不折叠」的用户;它和 /fold 共用 phase 1,保证两条路径产出一致。/health 返回 workers_ready / workers_total,供探活和「等就绪」用。请求还能指定 output_dir,让服务把全部扩散样本写到共享盘、客户端自己去读,而不是把好几个大 cif 塞进 HTTP 响应里。
多卡映射上有个小技巧值得一提:每个 worker 用 CUDA_VISIBLE_DEVICES 绑定一张物理卡,进程内部一律当「设备 0」来用。逻辑代码完全不必关心自己实际跑在第几张卡上。
6. gpuserver 的三条「对齐铁律」
把 client 的搜索接到 gpuserver 上时,出现过两类相当诡异的故障。一类是死锁:GPU 利用率挂在 0%,搜索永远卡住。另一类是 malloc(): corrupted top size,进程直接崩掉。
查到最后才明白,根因是 gpuserver 与 client 必须在三个维度上完全一致,否则不是死锁就是内存损坏:
| 必须对齐 | 不一致的后果 |
|---|---|
--max-seqs(每库的结果上限) | malloc(): corrupted top size,崩溃 |
--prefilter-mode | 行为不一致,甚至崩溃 |
CUDA_VISIBLE_DEVICES(连同设备字符串) | 死锁,GPU 0% |
实操上的关键是:gpuserver 的发现机制,是按 (库路径 + CUDA_VISIBLE_DEVICES 字符串) 做键的。所以一个库的 gpuserver 用什么 max-seqs 起的,client 去搜这个库时就得用同样的;起 gpuserver 的卡和搜它的 client,也必须看到同一组卡。最稳妥的做法,是把这套「每库参数表」在启动脚本里集中定义一次,让 server 和 client 共用同一份。
顺带一个教训:
corrupted top size这类报错极具误导性:它会把你往「分片有 bug」「线程问题」「二进制损坏」的方向带偏。但真正的根因,往往只是参数没对齐而已。
7. 性能数字(8×A800 实测)
把上面这些加速叠起来,以一个具体目标(PDB 7YV1)为例,实测数字如下:
| 场景 | MSA | 推理 | 总计 |
|---|---|---|---|
/fold 冷(新序列) | ~95 s | ~134 s | ~230 s |
/fold 暖(序列已缓存) | ~0 s | ~134 s | ~134 s |
/msa 冷 / 暖 | ~95 s / ~0 s | n/a | ~100 s / ~1 s |
几点观察。缓存是最大的杠杆:暖请求把整个 MSA 阶段抹掉,只剩推理。在批量场景里,一批输入共享大部分链,共享链命中缓存,只需搜真正变化的那一条。多卡则带来并行度:四个推理 worker 意味着一批请求最多四个同时折叠,每个占一张卡。容量方面,MSA 的 gpuserver 是并发安全的;UniProt 那张卡的峰值显存基本恒定(结果流式回主机,不随 batch 增大而堆显存),负载成本落在 CPU 的 result2msa,而不在 GPU。还有那块压不动的地板:冷 MSA 里的 result2msa(CPU)就是硬地板,约 80 到 90 秒,只能靠缓存(走暖路径)或降 max-seqs 来进一步压。
8. 几条可以带走的经验
- 常驻化的本质,是把一次性成本只付一次。 数据库、权重、环境都只付一次,之后每个请求只付增量。要不要常驻,判断标准很简单:看你的工作流是不是在反复折叠同类输入。
- 加速分层,各管一段。 常驻库省载入、align 上 GPU 省 CPU、批量加并发省串行、常驻权重又省载入、缓存省重复。只有理解每层的边界,才知道天花板在哪。在这套系统里,就是 CPU 上的
result2msa。 - 缓存粒度要对齐使用场景。 对配置固定的服务,缓存「成品 MSA」(序列级)比缓存「中间产物」(库级)更省也更直接。
- gpuserver 参数必须 server 与 client 对齐。
max-seqs、prefilter-mode、可见卡,三者一致;否则崩溃信息只会把你带偏。
归根结底,常驻加缓存这两招,把「对着同一个靶点反复折叠」的边际成本,从分钟级压到了「只剩推理」。对结构生物学里大量扫变体、做重设计的工作流来说,把折叠服务化是一笔很值的一次性投入。
致谢 / Credits
这篇文章本质上是把下面这些开源工作「服务化」的一次工程实践,核心模型与算法的功劳都归原作者:
- AlphaFast(RomeroLab):本文直接部署的对象。它把 MMseqs2-GPU 接进了 AF3 的数据流水线,带来了 MSA 的数量级提速;
gpuserver常驻、prefilter-mode 3、批量与并发搜索这些能力都来自它。 - AlphaFold3(Google DeepMind):底层的结构预测模型与数据流水线。使用时请遵守其权重与代码的相关条款。
- MMseqs2 / MMseqs2-GPU(Steinegger lab & Söding lab):GPU 加速的同源搜索引擎,是本文 MSA 提速的根基。
我自己的贡献仅在部署与服务化这一层:多卡角色的划分、常驻服务与缓存的设计、gpuserver 的对齐规则以及这些踩坑记录。模型与搜索算法本身的一切,都归功于上述项目。
参考文献
- Abramson, J., Adler, J., Dunger, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493-500 (2024). https://doi.org/10.1038/s41586-024-07487-w
- Steinegger, M. & Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nature Biotechnology 35, 1026-1028 (2017). https://doi.org/10.1038/nbt.3988
- Kallenborn, F., Chacón, A., Hundt, C., Sirelkhatim, H., Didi, K., Cha, S., Dallago, C., Mirdita, M., Schmidt, B. & Steinegger, M. GPU-accelerated homology search with MMseqs2. Nature Methods 22, 2024-2027 (2025). https://doi.org/10.1038/s41592-025-02819-8
- Perry, B.C., Kim, J. & Romero, P.A. AlphaFast: High-throughput AlphaFold 3 via GPU-accelerated MSA construction. bioRxiv (2026). https://doi.org/10.64898/2026.02.17.706409。代码:https://github.com/RomeroLab/alphafast
- Suzek, B.E., Wang, Y., Huang, H., McGarvey, P.B., Wu, C.H., the UniProt Consortium. UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 31(6), 926-932 (2015). https://doi.org/10.1093/bioinformatics/btu739
- Richardson, L., Allen, B., Baldi, G. et al. MGnify: the microbiome sequence data analysis resource in 2023. Nucleic Acids Research 51(D1), D753-D759 (2023). https://doi.org/10.1093/nar/gkac1080
- Steinegger, M., Mirdita, M. & Söding, J. Protein-level assembly increases protein sequence recovery from metagenomic samples manyfold. Nature Methods 16, 603-606 (2019). https://doi.org/10.1038/s41592-019-0437-4(Big Fantastic Database,BFD)
- The UniProt Consortium. UniProt: the Universal Protein Knowledgebase in 2023. Nucleic Acids Research 51(D1), D523-D531 (2023). https://doi.org/10.1093/nar/gkac1052